Лечение астроцитомы головного мозга
Астроцитома — самая распространенная и одна из самых агрессивных разновидностей опухолей головного мозга. Новообразование оказывается злокачественным в 50% выявленных случаев. Пациентам, которым подтвердили доброкачественность опухоли, важно немедленно начать лечении. Если своевременно не пройти терапию, астроцитома с большой вероятностью может перерасти в злокачественную. знайте больше информации о методах борьбы с заболеванием и прогнозах – читайте статью MediGlobus.
Слушать статью:
Клиническая картина
Клиническая картина зависит от локализации и степени злокачественности астроцитомы (подробно клинику см. Головной мозг, опухоли).
Диагноз ставится на основании общемозговых и очаговых симптомов развития внутримозговой опухоли (см. Головной мозг, опухоли). До операции обычно ограничиваются предположением о наличии глиомы, соответственно тяжести и динамике симптомов — доброкачественной или злокачественной. Астроцитарная природа глиомы нередко очевидна для нейрохирурга уже во время операции на основании макроскопических данных. Точный диагноз астроцитома устанавливается гистологическим исследованием.
Причины развития астроцитомы
Точные причины развития астроцитомы не установлены. Сегодня можно назвать только предрасполагающие факторы, способствующие развитию болезни:
- наследственная предрасположенность к развитию онкологических заболеваний. Риск развития заболевания повышен при наличии у пациента нейрофиброматоза или бугорчатого склероза;
- воздействие неблагоприятных факторов окружающей среды – ионизирующего излучения, канцерогенных веществ, находящихся в воде воздухе;
- профессиональные вредности – при воздействии на человека токсических химических соединений, например, продуктов нефтепереработки, физических факторов;
- вирусные заболевания – некоторые вирусы обладают высокой онкогенностью и способствуют развитию раковых опухолей.
Новые технологии лечения
Ведущим методом борьбы с астроцитомой является ее максимально полное удаление, насколько позволяет локализация новообразования. Для этого в крупных иностранных клиниках применяются новые технологии:
- нейронавигация. На основании данных предоперационной МРТ и компьютерных программ создают трехмерные графические модели. Пространственная реконструкция мозга и находящейся внутри него опухоли используется в ходе ее удаления для позиционирования инструментов в режиме реального времени. Это позволяет не только наиболее полно иссечь опухоль и обеспечить к ней максимально бережный доступ. В работе нейронавигационной системы непрерывно обрабатываются и анализируются данные нескольких видов визуализационных и функциональных исследований, которые проводятся интраоперационно. В результате в каждый момент времени хирург получает четкое представление о структурных и функциональных изменениях в операционном поле и быстро реагирует на них;
- маркировка опухоли. Перед операцией пациент принимает внутрь препарат, который накапливают клетки опухоли. В ходе операции, под определенным освещением, они светятся розовым цветом. В головном мозге, где каждый миллиметр ткани незаменим, такая методика, обеспечивающая высочайшую точность действий хирурга, особенно важна;
- Awake craniotomy, дословно – «Бодрствующая краниотомия». Ткань мозга безболезненна, поэтому после доступа к нему под общей или местной анестезией через кости черепа и оболочки мозга дальнейшего обезболивания не требуется. На этом этапе выполняют корковое картографирование. Для этого стимулируют определенные зоны мозга электродом и наблюдают реакцию пациента. По изменению его движений, чувствительности, речи уточняют расположение особо функционально значимых участков мозга и избегают их травмирования при удалении опухоли.
Доброкачественные астроцитомы, которые можно удалить тотально, применения после операции других методов лечения не требуют. Пациенты, страдающие астроцитомами высокой степени злокачественности, получают после операции химиотерапию и радиотерапию.
Если операция противопоказана, применяются радиохирургические методы Гамма-нож и Кибер-нож, позволяющие доставить с высочайшей точностью фокусированное высокодозное излучение в опухоль. Под его действием повреждается ДНК опухолевых клеток, и они теряют способность к росту и размножению. В итоге очаг постепенно уменьшается. Иногда эти методы сочетают с хирургической операцией, если опухоль невозможно было удалить радикально. Существуют также варианты внутримозговой брахитерапии, когда в ложе удаленной опухоли или в ее толщу (при невозможности резекции) помещают источник радиоактивного излучения, действующий только в пределах новообразования.
Химиотерапия проводится как до, так и после операции. Если в программу лечения включена лучевая терапия, химиопрепараты назначаются через две недели после ее окончания. Это комбинации прокарбазина, ломустина, винкристина или других средств. Состав протокола, продолжительность и количество курсов определяются в каждом случае индивидуально.
Обычной практикой ведущих нейрохирургических центров мира является лечение астроцитом с использованием нейронавигации и системы Awake craniotomy для хирургического удаления опухоли. Широко применяются радиохирургические методы Гамма-нож и Кибер-нож, комплексная химиотерапия, иммунные и таргетные препараты.
Симптомы и клинические проявления
При астроцитоме возникают признаки общего типа, а также местные, соответствующие тому отделу головного мозга, где расположилась опухоль. Большинство симптомов вызвано повышением черепного давления или интоксикацией организма от пораженных клеток. На первых этапах развитие астроцитомы может протекать незаметно, что усложняет ее своевременное выявление.
- головные боли ноющего характера;
- ухудшение аппетита;
- тошнота, рвота;
- медлительность речи;
- нарушения внимания и памяти;
- двоение или туман в глазах;
- предобморочное состояние;
- усталость, общее недомогание;
- проблемы с координацией;
- судороги, эпилептические припадки;
- перепады настроения.
Где и как проявляется опухоль:
- Лобная доля. Главным отличием астроцитомы, локализованной на этом участке, считаются психопатологические симптомы. Больной может испытывать эйфорию, равнодушно относиться к заболеванию, проявлять агрессию по отношению к окружающим. Постепенно психика разрушается полностью. Также могут возникнуть нарушения памяти и мышления или моторные нарушения речи. Возможен паралич конечностей.
- снижение интеллекта;
- ухудшение памяти;
- приступы агрессии и выраженного психического возбуждения; снижение мотивации, апатия, инертность;
- выраженная общая слабость.
- Височные доли. Больной часто испытывает галлюцинации слухового, зрительного или вкусового характера. С развитием астроцитомы такие проявления становятся эффектом, предвещающим скорый эпилептический приступ с потерей сознания. Часто также появляются нарушения речи сенсорного типа и слуховая агнозия, из-за чего человек перестает понимать даже написанные слова и распознавать звуки. Глиобластома или анапластическая астроцитома головного мозга при такой локализации часто приводят к быстрому летальному исходу.
- Теменная доля. Больные с таким расположением астроцитомы испытывают проблемы с узнаванием предметов на ощупь, невозможностью контролировать конечности при выполнении целенаправленных действий, эпилептическими припадками. Иногда возникают нарушения речи, письма или счета.
- Затылочная доля. Основным признаком развития астроцитомы на этом участке считаются зрительные галлюцинации. Человек может видеть то, чего нет, или реальные предметы могут изменять в его глазах свой внешний вид и размер. Возможно частичное выпадение поля зрения из обоих глаз.
- Если будет поражен мозжечок, то могут возникать проблемы с походкой и координацией движения. Поврежденный желудочек приводит к рефлекторному напряжению шеи с изменением положения головы. Пораженные продолговатый и спинной мозг могут вызвать проблемы с конечностями и ходьбой.
Общие симптомы
Головная боль, тошнота и рвота
Симптомы опухоли мозжечка, как на ранних стадиях, так и на более поздних зачастую включают головную боль, тошноту и рвоту. По мере роста новообразования, оно блокирует жидкость, окружающую мозг. Это приводит к гидроцефалии или повышенному содержанию жидкости в черепе. Избыточное содержание жидкости в черепной коробке влечет повышенное давление в нем, что вызывает тошноту и приступы головной боли. У некоторых людей головная боль после пробуждения может быть чрезвычайно сильной и приводить к рвоте. При этом обычные средства от головной боли не имеют существенного действия на больного. По ходу дня боль зачастую спадает.
Нарушение походки
Мозжечок контролирует координацию мускулов. По мере роста новообразований этого органа повышенное внутричерепное давление может нарушать его функции. Порой развиваться неуклюжесть и нарушается координация движений. Симптомы опухоли мозжечка приводят к изменению походки, она становится покачивающейся и шаткой.
Повреждение черепных нервов
Задняя черепная ямка характеризуется небольшим объемом. В некоторых случаях новообразование по мере роста заполняет ее пространство и может повредить прилежащие структуры, например, черепные нервы. Травмирование нервов приводит к расширению зрачков, потере периферического зрения, отклонению глазного яблока и размытому зрению. У некоторых развивается ослабление мимических мышц, Нарушение чувствительности участков лица, потеря слуха и нарушение вкусового восприятия.
ТУБЕРОЗНЫЙ СКЛЕРОЗ
Туберозный склероз — генетически детерминированное заболевание, относится к группе нейроэктодермальных нарушений, характеризуется поражением нервной системы, кожи и наличием доброкачественных опухолей (гамартом) в различных органах. Первое описание кли
Туберозный склероз — генетически детерминированное заболевание, относится к группе нейроэктодермальных нарушений, характеризуется поражением нервной системы, кожи и наличием доброкачественных опухолей (гамартом) в различных органах.
Первое описание клинического случая было сделано в 1862 г. F. von Recklinghausen. В 1880 г. D.-M. Bourneville подробно описал изменения, возникающие в головном мозге при этом заболевании [1, 24].
Частота туберозного склероза составляет 1 : 30 000 населения. Распространенность среди новорожденных варьирует от 1 : 6000 до 1:10 000 [39].
Туберозный склероз наследуется по аутосомно-доминантному типу. Большинство случаев заболевания (80%) является следствием мутации de novo. Болезнь отличается варьирующей экспрессивностью и почти 100%-ной пенетрантностью. Развитие туберозного склероза определяется двумя генами, локализованными в участке 34 длинного плеча 9-й хромосомы (туберозный склероз 1-го типа — TSC1, кодирует белок гамартин) и в участке 13 короткого плеча 16-й хромосомы (туберозный склероз 2-го типа — TSC2, кодирует белок туберин) [32].
В 1998 г. были приняты диагностические критерии заболевания (табл.). Несомненный диагноз туберозного склероза: два или один первичный признак + два вторичных признака. Возможный диагноз: один первичный признак + один вторичный признак. Предположительный диагноз: или один первичный признак, или два (и больше) вторичных признака.
Клиническая характеристика
Кожные изменения при туберозном склерозе представлены гипопигментными пятнами, ангиофибромами лица, участками «шагреневой кожи», околоногтевыми фибромами, фиброзными бляшками, белыми прядями волос [43].
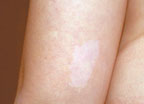 |
| Рисунок 1. Гипопигментное пятно |
Гипопигментные пятна являются одним из наиболее частых кожных проявлений туберозного склероза (рис. 1). Они встречаются в 90% случаев и нередко обнаруживаются с рождения, являясь одним из манифестных признаков заболевания. С возрастом наблюдается тенденция к увеличению их числа. Гипопигментные пятна при туберозном склерозе локализуются преимущественно на туловище и ягодицах. Характерной их особенностью является асимметричность расположения. Отмечена вариабельность числа, размера и формы пятен.
С младенчества могут выявляться белые пряди волос, ресниц и бровей, которые, как и гипопигментные пятна, являются характерным признаком туберозного склероза.
Наряду с гипопигментными пятнами при туберозном склерозе в 15,4% случаев встречаются пигментные пятна цвета «кофе с молоком», что не превышает средних популяционных значений.
Ангиофибромы лица — облигатный признак туберозного склероза — наблюдаются в 47–90% случаев и появляются, как правило, после 4 лет. Внешне они представляют собой папулы или узлы розового или красного цвета с гладкой, блестящей поверхностью. Ангиофибромы располагаются на лице симметрично, с двух сторон — на щеках и на носу по типу «крыльев бабочки», а также на подбородке (рис. 2).
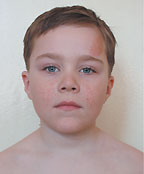 |
| Рисунок 2. Ангиофиброма лица |
«Шагреневая кожа» (peau chagrin в переводе с франц. — «недубленая, грубая, жесткая кожа») — облигатный признак туберозного склероза, встречается в 21–68% случаев. В большинстве случаев «шагреневая кожа» появляется на втором десятилетии жизни. Участки «шагреневой кожи» наблюдаются преимущественно в пояснично-крестцовой области, имеют плотную консистенцию, желтовато-коричневый или розовый цвет, умеренно выступают над поверхностью окружающей кожи. Количество участков «шагреневой кожи» вариабельно, но чаще они бывают единичными. Размер их колеблется от нескольких миллиметров до 10 см и более.
Фиброзные бляшки встречаются у 25% больных с туберозным склерозом и также являются облигатным признаком заболевания. Фиброзные бляшки имеют бежевый цвет, шероховаты на ощупь и несколько выступают над окружающей кожей. Они часто появляются уже на первом году жизни и являются, таким образом, одним из первых клинических симптомов заболевания. Чаще всего фиброзные бляшки локализуются на лбу. Размеры и число бляшек могут варьировать.
 |
| Рисунок 3. Околоногтевые фибромы |
Околоногтевые фибромы — облигатный признак туберозного склероза (рис. 3). Они представляют собой тусклые, красные либо мясного цвета папулы или узлы, растущие от ногтевого ложа или вокруг ногтевой пластинки. Околоногтевые фибромы встречаются в 17–52% случаев. В большинстве случаев околоногтевые фибромы появляются на втором десятилетии жизни. Наиболее часто они локализуются на ногах. Размер их варьирует от 1 мм до 1 см в диаметре. Наличие околоногтевых фибром более характерно для женщин.
Мягкие фибромы встречаются у 30% больных. Они представляют собой множественные или единичные мягкие образования на ножках, мешотчатой формы, растущие на шее, туловище и конечностях (molluscum fibrosum pendulum). Другой вариант мягких фибром представляет собой множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования, размером меньше булавочной головки, располагающиеся на туловище и шее и напоминающие гусиную кожу.
Наиболее типичными поражениями головного мозга при туберозном склерозе являются корковые туберы, субэпендимальные узлы и аномалии белого вещества мозга [20, 29].
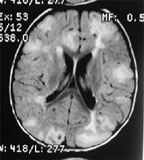 |
| Рисунок 4. Магнитно-резонансное исследование головного мозга |
Корковые туберы различаются по своим размерам, локализации, консистенции и форме. Размер корковых туберов варьирует от нескольких миллиметров до нескольких сантиметров. Корковые туберы располагаются в виде выступов над единичной или прилегающими бороздами. Они расширяют борозду и сглаживают грань между серым и белым веществом. Туберы могут быть как единичными, так и множественными, имеют диффузную локализацию. Кальцификация туберов отмечается в 54% случаев и увеличивается с возрастом больных.
Своевременное выявление корковых туберов и кальцификатов мозга очень важно для диагностики туберозного склероза. Наибольшую значимость в верификации туберов при обследовании больных имеет магнитно-резонансная томография (МРТ), которая позволяет визуализировать туберы в 95% случаев (рис. 4).
 |
| Рисунок 5. Компьютерно-томографическое исследование головного мозга |
Субэпендимальные узлы встречаются в 95% случаев и выявляются как при компьютерном томографическом (КТ), так и при МРТ-исследованиях мозга. Субэпендимальные узлы в большинстве случаев множественные, прилегающие друг к другу. Локализуются, как правило, в стенках боковых желудочков, реже — в стенках III и IV желудочков мозга. При локализации в стенках боковых желудочков субэпендимальные узлы глубинной частью могут внедряться в хвостатое ядро или таламус. Форма субэпендимальных узлов обычно округлая или вытянутая. По мере роста ребенка в субэпендимальных узлах происходит постепенное отложение кальция. На компьютерных томограммах доминирующим признаком заболевания являются множественные, полностью или частично кальцифицированные, субэпендимальные узлы округлой формы, локализующиеся в стенках боковых желудочков (рис. 5). КТ-исследование более чувствительно, когда речь идет о выявлении кальцифицированных субэпендимальных узлов.
Субэпендимальные узлы нередко трансформируются в гигантоклеточную астроцитому и выявляются у 10–15% больных [47, 51]. Субэпендимальные гигантоклеточные астроцитомы манифестируют обычно между 5 и 10 годами жизни (средний возраст в момент выявления опухоли — 13 лет), как правило, имеют тенденцию к росту и всегда локализуются у межжелудочкового отверстия. Для диагностики гигантоклеточных астроцитом применяются как КТ-, так и МРТ-исследования (рис. 6). В клинической практике нередко используют оба нейрофизиологических метода.
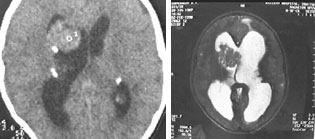 |
| Рисунок 6. Гигантоклеточная астроцитома: a — КТ-исследование; б — МРТ-исследование |
Учитывая тот факт, что при туберозном склерозе гигантоклеточные астроцитомы наблюдаются относительно часто, рекомендуется динамическое проведение нейрорадиологических исследований у больных детей, не реже 1 раза в 2 года. В случае появления таких клинических симптомов, как головная боль, рвота, ухудшение зрения, необходимо экстренное проведение нейрорадиологических исследований.
Поражение белого вещества головного мозга при туберозном склерозе характеризуется появлением своеобразных островков, состоящих из групп гетеротопических кластерных клеток и располагающихся вдоль линий, соединяющих эпендиму стенок желудочков и туберы. Данные линии соответствуют нормальным миграционным путям спонгиобластов во время эмбриогенеза.
У 10% больных при туберозном склерозе описаны поражения мозжечка.
Поражения нервной системы являются доминирующими в клинической картине туберозного склероза. Наиболее характерны судорожные пароксизмы, умственная отсталость, нарушения поведения, изменения в цикле «сон–бодрствование».
Судорожные пароксизмы — один из наиболее значимых симптомов туберозного склероза — наблюдаются у 80–92% больных [19, 38] и чаще всего являются манифестным симптомом заболевания. Эпилептические пароксизмы при туберозном склерозе нередко резистентны к противосудорожной терапии, могут приводить к развитию нарушений интеллекта и поведения и являются одной из главных причин инвалидности у детей с туберозным склерозом. P. Curatolo [17] отмечено, что среди факторов, детерминирующих резистентность к противосудорожной терапии, наибольшее значение имеют: дебют в возрасте до 1 года, наличие нескольких типов приступов, высокая частота приступов, изменение характера приступов с течением заболевания.
Умственная отсталость при туберозном склерозе наблюдается в 48% случаев [10, 18, 28] и варьирует от умеренной до глубокой степени. Одной из основных причин, которые определяют ее возникновение, являются судороги, возникающие на первом году жизни. Нарушение интеллекта при туберозном склерозе сочетается с изменениями поведения в виде аутизма, гиперактивности, агрессивности.
При туберозном склерозе лечение преимущественно носит симптоматический характер. Подбор антиконвульсантов проводится с учетом характера судорожных приступов. Правильно подобранная терапия позволяет избежать возникновения осложнений, характерных при эпилепсии. Для доказательства неэффективности препарата длительность его применения в максимально переносимой дозе (максимальная доза препарата, которую переносит больной без появления побочных эффектов) должна быть равна 5–6 межприступным интервалам. Как правило, больные с туберозным склерозом из-за резистентности приступов к проводимой терапии вынуждены принимать два противосудорожных препарата.
Согласно международным критериям [3, 13], резистентной называется эпилепсия, при которой неэффективны:
- последовательная монотерапия двумя антиконвульсантами первого выбора (карбамазепин, вальпроат натрия) в максимально переносимах дозах;
- комбинация двух антиконвульсантов первого выбора в максимально переносимых дозах;
- антиконвульсанты нового поколения (ламотриджин, вигабатрин, тиагабин) с широким спектром действия, которые обладают, в отличие от карбамазепинов и вальпроатов, иным механизмом действия.
Лечение поражений головного мозга
Принципиально важным является подбор антиконвульсантов с учетом характера эпилептических приступов. Для лечения инфантильных спазмов на фоне туберозного склероза, препаратами первого выбора являются вигабатрин (сабрил, сабрилекс) и гормональные средства (синактен депо — синтетический полипептид, обладающий свойствами эндогенного адренокортикотропного гормона). Вигабатрин усиливает ГАМКергическое ингибирование посредством необратимой блокады ГАМК-трансаминазы. Способ применения — пероральный (таблетки, порошок). Кратность приема — 1–2 раза в день. Начальная доза — 40 мг/кг. Поддерживающая доза — 80–100 мг/кг в сутки. Влияние на другие противосудорожные препараты клинически не проявляется. Основные побочные эффекты: дозозависимые — гиперактивность, сонливость; хронические — прибавка массы тела, концентрическое сужение полей зрения [52].
Синактен депо влияет на процессы регуляции судорожной активности головного мозга. При применении адренокортикотропного гормона (АКТГ) модифицируется образование различных биогенных аминов, включая серотонин, увеличивается уровень GABA и рецепторная аффинность, усиливается проницаемость гематоэнцефалического барьера. В настоящее время рекомендуются два варианта курсовой терапии: короткий — 3–8 нед, либо продолжительный — 4–6 мес. Наиболее типичными побочными эффектами при назначении АКТГ являются возбуждение, нарушение сна, эндокринные и электролитные нарушения, понижение сопротивляемости организма к инфекциям, эрозивные изменения слизистой оболочки желудка и кишечника, гиперпигментация кожи. Лечение инфантильных спазмов кортикотропными препаратами при туберозном склерозе приводит к значительному росту рабдомиом сердца [27].
Средствами второго выбора для лечения инфантильных спазмов на фоне туберозного склероза являются препараты вальпроевой кислоты (конвулекс — капли, сироп, таблетки обычного и пролонгированного действия, депакин – сироп, таблетки обычного и пролонгированного действия). Их доза составляет 40–100 мг/кг в сут.
Препаратами первого выбора для лечения парциальных эпилептических пароксизмов являются базовые антиконвульсанты — вальпроаты (конвулекс пролонгированного действия, депакин хроно) и карбамазепин (тегретол, финлепсин и их ретардированные формы).
Препараты вальпроевой кислоты усиливают ГАМКергическое ингибирование и уменьшают пароксизмальные разряды нейронов (блокада Na + — и Ca + -каналов Т-типа). Способ применения — пероральный (таблетки, таблетки пролонгированного действия, капли, сироп). Применяются в дозе 15–40 мг/кг в сутки. Кратность приема — 1–3 раза в день в зависимости от формы выпуска. При комбинации с вальпроатами увеличивается концентрация в крови ламотриджина, карбамазепина, фенобарбитала, фенитоина, фелбамата, этосуксимида, в связи с чем их доза должна быть уменьшена. Основные побочные эффекты: идиосинкразия — панкреатит, печеночная недостаточность; дозозависимые — сонливость, тошнота, рвота, атаксия, тремор, облысение; хронические — прибавка в весе, тромбоцитопения, нарушения менструального цикла, поликистоз яичников.
Карбамазепины уменьшают пароксизмальные разряды нейронов (блокада Na + -каналов Т-типа) и ослабляют влияние возбуждающих аминокислот — глутамата), индуцируют микросомальные ферменты печени, ускоряя тем самым собственный метаболизм (аутоиндукция). Способ применения — пероральный (таблетки, таблетки пролонгированного действия). Применяются в дозе 15–30 мг/кг в сутки. При комбинации с вальпроатами их концентрация в крови снижается. Основные побочные эффекты: идиосинкразия — лейкопения, тромбоцитопения, сыпь, гиперчувствительность с развитием гепатотоксичности; дозозависимые — сонливость, головная боль, головокружение, диплопия, нистагм, тошнота, рвота, запоры; хронические — гипонатриемия, недостаточность фолиевой кислоты. Карбамазепины, применяемые для лечения эпилепсии у больных с туберозным склерозом и рабдомиомой сердца, могут обусловливать поперечный атриовентрикулярный блок [53].
В случае отсутствия эффекта от монотерапии, возможно сочетание вальпроатов и карбамазепина с ламотриджином (ламикталом). Ламотриджин уменьшает пароксизмальные разряды нейронов — блокада Na + -каналов Т-типа и ослабляет возбуждающее влияние глутамата. Способ применения — пероральный. Кратность приема — 1–2 раза в день. Начальная доза — 0,5 мг/кг (при комбинации с вальпроатами — 0,2 мг/кг). Поддерживающая доза — 2–10 мг/кг (при комбинации с вальпроатами — 4–12 мг/кг). Доза повышается медленно, в течение 12 нед. При комбинации с вальпроатами их концентрация в крови возрастает. Основные побочные действия: идиосинкразия — синдром Стивена-Джонсона, синдром Лайелла; дозозависимые — сонливость, аллергические реакции, сыпь, рвота, атаксия, головная боль, гиперкинезия.
При резистентных формах эпилепсий судорожные приступы могут сохраняться, несмотря на проводимое лечение различными противосудорожными препаратами и их комбинациями [4]. Один из путей решения данной проблемы — применение новых антиконвульсантов. Новые препараты должны быть высокоэффективными, иметь широкий спектр терапевтического действия и минимум побочных эффектов, не вызывать аггравации приступов. Топирамат (топамакс) и леветирацетам (кеппра) представляют собой современные лекарственные средства, отвечающие данным требованиям.
Топирамат (топамакс) рекомендуется к применению как препарат первого выбора при парциальных и генерализованных судорожных приступах, а также в комбинации с другими препаратами [2, 5].
Топирамат представляет собой сульфатозамещенный моносахарид. Препарат обладает сложным и множественным механизмом действия и воздействует на различные звенья патогенеза эпилепсии: ослабляет (глутамат) возбуждающее и усиливает (ГАМК) тормозное влияние. Стабилизирует ионные (натриевые и кальциевые) каналы мембран, угнетает активность карбоангидразы. Для топирамата характерны антиэксайтотоксические (препятствует дегенерации двигательных нейронов) и нейропротективные свойства (снижает вероятность осложнений при фокальной ишемии мозга).
Фармакокинетический профиль топирамата характеризуется быстрой и практически полной абсорбцией, не зависящей от приема пищи; линейной кинетикой, что позволяет предсказать изменения концентрации препарата в плазме крови с изменением дозы препарата или режима его введения (постоянная концентрация препарата достигается через 4 дня). Топирамат не обладает аутоиндукцией и активными метаболитами, мало связывается с белками плазмы, что ограничивает его взаимодействие с другими препаратами, выводится в основном почками, обладает длительным периодом полувыведения — 19–23 ч, при сочетании с препаратами-индукторами белков плазмы — 12–15 ч. В сочетании с топираматом концентрация препаратов вальпроевой кислоты в плазме крови снижается минимально (11%), концентрация карбамазепина и его метаболитов остается прежней, тогда как концентрация фенитоина может возрастать. В свою очередь, карбамазепин и фенитоин несколько снижают концентрацию топирамата в крови.
У больных с патологией почек доза препарата должна быть уменьшена, у новорожденных и детей требуются более высокие дозы препарата на килограмм веса в связи с большей скоростью выведения. Способ применения — пероральный. Кратность приема — 2 раза в день, независимо от приема пищи. Доза препарата — 1–20 мг/кг в сутки (средняя терапевтическая доза — 6–10 мг/кг в сутки). Доза титруется медленно — еженедельно увеличивается на 25 мг до достижения терапевтического эффекта. Топамакс обладает благоприятным профилем безопасности у пациентов различных возрастных групп. Побочные эффекты оцениваются как легкие и умеренные и носят преходящий характер; наблюдаются они в начале курса терапии, на этапе титрования. Основные побочные действия — сонливость, нарушение концентрации внимания, памяти, головная боль, парестезии. При длительной терапии возможно снижение массы тела, связанное с ухудшением аппетита [7, 23, 46].
Леветирацетам (кеппра) применяется как в монотерапии, так и в комбинации с другими противосудорожными препаратами для лечения парциальных и генерализованных форм эпилепсий [8, 14, 16, 49].
Леветирацетам по химической структуре подобен пирацетаму. Точный механизм противосудорожного действия неизвестен. Препарат обладает высокой биодоступностью, линейной фармакокинетикой, мало связывается с белками, не влияет на печеночный метаболизм, обладает низким потенциалом лекарственных взаимодействий, не требует длительного титрования. Дозы препарата — 10–60 мг/кг в сутки (у детей с резистентными формами эпилепсий — до 100 мг/кг в сутки). Способ применения — пероральный. Кратность приема — 2 раза в день, независимо от приема пищи. Побочные эффекты леветирацетама легкие и крайне редкие — сонливость, астения, головная боль, головокружение, депрессия, тревога, эмоциональная лабильность.
Одной из самых сложных проблем при лечении больных с туберозным склерозом является коррекция умственной отсталости. В связи с наличием судорожных пароксизмов применение ноотропных препаратов и стимулирующей терапии во многих случаях противопоказано. Основной акцент при работе с умственно отсталыми пациентами делается на нейропсихологической реабилитации.
При появлении гигантоклеточной астроцитомы тактика ведения больных, как правило, выжидательная. Хирургическое вмешательство показано лишь в случае быстрого роста опухоли, вызывающего повышение внутричерепного давления.
Поражение органов зрения при туберозном склерозе характеризуется появлением гамартом сетчатки и зрительного нерва, которые выявляются примерно у 50% больных. У половины больных они множественные. Выделяют три наиболее типичных варианта гамартом сетчатки. При первом, наиболее распространенном варианте гамартомы имеют нежную, относительно плоскую и гладкую поверхность, оранжево-розовый цвет, округлую или овальную форму, локализуются преимущественно в поверхностных слоях сетчатки. При втором — гамартомы имеют узловатый вид и напоминают тутовую ягоду. Они белого цвета, кальцифицированные, светонепроницаемые. При третьем варианте гамартомы сочетают в себе признаки первых двух. Они имеют округлую форму с узловатым и кальцифицированным центром и полупрозрачной, гладкой периферией оранжево-розового цвета (рис. 7).
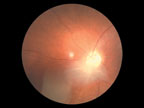 |
| Рисунок 7. Гамартома диска зрительного нерва |
Клинические проявления гамартом наблюдаются крайне редко. Основным симптомом является прогрессирующее снижение зрения [42].
Изменения сердечно-сосудистой системы при туберозном склерозе проявляются развитием рабдомиом, которые нередко служат первым клиническим признаком туберозного склероза наряду с гипопигментными пятнами. Рабдомиомы встречаются в 30–60 % случаев и выявляются чаще у лиц мужского пола (соотношение 2 : 1). Наиболее высокая частота рабдомиомы сердца при туберозном склерозе наблюдается у новорожденных (у 21 из 23 детей) и детей грудного возраста (у 11 из 33 детей) [30, 31, 34].
Рабдомиоматозные образования могут быть в виде одного узла или множественными. Они, как правило, локализуются в желудочках и имеют смешанный интраэкстрамуральный рост.
В очень редких случаях рабдомиомы могут локализоваться в предсердиях, исходя из межпредсердной перегородки. Рабдомиомы различаются по своим форме и размерам, которые варьируют от нескольких миллиметров до нескольких сантиметров. Опухоли имеют неправильную форму и всегда четко отделены от окружающих тканей [35, 36] (рис. 8).
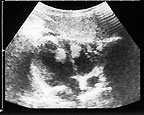 |
| Рисунок 8. Рабдомиомы сердца |
Ультразвуковое исследование позволяет выявить опухоль сердца еще во время внутриутробного развития плода, начиная с 21 нед гестации. Во всех случаях внутриутробной диагностики опухоли у новорожденного должен быть исключен туберозный склероз даже при отсутствии семейного анамнеза [21].
МРТ-исследование сердца более информативно при определении степени прорастания миокарда, поскольку позволяет определить «демаркационную» линию опухоли, отделяющую от рабочего миокарда [9].
Клинические симптомы рабдомиом у новорожденных различны. При опухолях, диагностированных внутриутробно, в 4 случаях из 11 наблюдалась внутриутробная смерть плода [26]. Примерно у 50% новорожденных опухоль может выявиться случайно при проведении планового эхокардиографического обследования по поводу туберозного склероза. Обычно эти опухоли не нарушают гемодинамику и не имеют выраженного интрамурального роста. Известны случаи диагностики рабдомиомы при обследовании новорожденных по поводу пароксизмальной тахикардии [12].
При массивных опухолях может наблюдаться внутриутробная смерть плода либо дети рождаются преждевременно с низкой оценкой по шкале Апгар, имеют распространенные отеки и выраженный цианоз. Имеются сообщения о случаях смерти новорожденных от застойной сердечной недостаточности [44].
Замечено, что рабдомиомы сердца, как правило, быстро увеличиваются во время второй половины беременности, в основном достигают максимальных величин к моменту рождения, а затем постепенно уменьшаются в размерах. Большинство рабдомиом исчезают бесследно. Спонтанная регрессия рабдомиом наблюдалась у детей младше 6 лет. После 6 лет опухоли обычно не исчезают, однако могут несколько уменьшаться в размере [22, 36]. Регресс опухолей может наблюдаться как в размере, так и в числе.
Детальное изучение кардиологических нарушений при туберозном склерозе свидетельствует о высокой частоте не только опухолей сердца, но и различных нарушений ритма и проводимости, которые могут приводить к внезапной смерти. В периоде новорожденности рабдомиомы сопровождаются нарушениями ритма сердца у 13 из 33 детей, в том числе синдромом Вольфа–Паркинсона–Уайта и пароксизмальной тахикардией [11, 37, 40]. У детей с туберозным склерозом старшего возраста рабдомиомы сердца обычно бессимптомны. Редко могут наблюдаться преходящая блокада левой ножки пучка Гиса, псевдоишемические изменения на электрокардиограмме.
Хирургическое лечение показано, если опухоль вызывает обструкцию в приточных или отточных отделах желудочков или обусловливает тяжелые нарушения ритма сердца [6].
Изменения органов дыхания встречаются у 1% больных c туберозным склерозом (поражаются в основном женщины). Легкие вовлекаются в патологический процесс после 30 лет. Наиболее типичным поражением легких являются кисты. Первые клинические симптомы — это дыхательная недостаточность и рецидивирующий пневмоторакс. На рентгенограмме грудной клетки выявляется усиленный рисунок легочной паренхимы и характерный паттерн «сотовых» легких, распространяющийся на всю их паренхиму или только на ее изолированные участки [33].
Изменения в органах желудочно-кишечного тракта при туберозном склерозе отличаются разнообразием, встречаются относительно часто и проявляются патологией ротовой полости, печени и прямой кишки [25].
Наиболее типичные нарушения, выявляемые при исследовании ротовой полости, — это узловые опухоли, фибромы или папилломы. Они локализуются главным образом на переднем крае десен, преимущественно на верхней челюсти, но также встречаются на губах, слизистой оболочке щек, спинке языка и небе. Дефекты эмали зубов отмечаются практически у всех больных с туберозным склерозом. Одним из наиболее типичных нарушений является дефект эмали зубов в виде углублений, число которых варьирует от 1 до 11 на каждом зубе. Возможны несколько вариантов дефектов эмали зубов: небольшие углубления, невидимые без увеличения, около 4 мкм в диаметре; углубления в эмали зубов до 60 мкм в диаметре; кратерообразные углубления в эмали зубов, видимые невооруженным глазом, около 100 мкм в диаметре.
При туберозном склерозе в печени появляются одиночные и множественные гамартомы и ангиомиолипомы, наблюдаемые у 10% больных.
Изменения в кишечнике при туберозном склерозе проявляются главным образом ректальными полипами, которые встречаются в 50–78% случаев. Ректальные полипы, как правило, выявляются у больных старше 20 лет. Клинически они бессимптомны и лишь в отдельных случаях возможны боли при дефекации. Ректальные полипы выявляются при пальцевом исследования прямой кишки и с помощью инструментальных методов исследования (сигмоидоскопии, колоноскопии, контрастной рентгенографии прямой кишки).
Полипы при туберозном склерозе обычно имеют благоприятный прогноз.
Наиболее типичной патологией почек при туберозном склерозе являются ангиомиолипомы и кисты, которые чаще всего бывают множественными и двусторонними; однако встречаются и единичные кисты и ангиомиолипомы с локализацией только в одной почке. По данным разных авторов, поражение почек при туберозном склерозе встречается в 47–85% случаев, причем изолированное поражение почек в дебюте заболевания встречается в 1–2%, а при аутопсийном исследовании, по данным клиники Mayo, в 100% случаев туберозного склероза. При этом ангиомиолипомы выявлены у 48% больных, кисты — у 35% и сочетание ангиомиолипом и кист — у 17% пациентов [50]. Реже встречаются и другие виды опухолей, такие, как почечноклеточная карцинома (в 5%), онкоцитома, а также неопухолевое поражение почек — фокальный сегментарный гломерулосклероз. Описаны также сосудистые дисплазии и пороки почечной ткани, тубулоинтерстициальный нефрит, мембранопролиферативный гломерулонефрит и нефрокальциноз. Как правило, поражение почек манифестирует во 2–3-й декадах жизни, однако клинические симптомы могут проявляться и в более ранние сроки.
Ангиомиолипомы при туберозном склерозе являются одним из главных диагностических признаков, при условии множественности, а также при двустороннем характере поражения, в то время как спорадические ангиомиолипомы, не связанные с туберозным склерозом, бывают единичными и односторонними. Ангиомиолипомы представляют собой доброкачественные образования, состоящие из аномальных сосудов, гладкомышечных клеток и жировой ткани с локально-инвазивным ростом. Соотношение этих компонентов в каждом отдельном случае различно.
При ультразвуковом исследовании почек ангиомиолипомы выглядят как эхопозитивные округлые образования в почечной паренхиме. При величине их менее 4 см в диаметре, как правило, никаких клинических проявлений не отмечается. При дальнейшем росте в центре ангиомиолипомы может появиться участок просветления, свидетельствующий о потенциальной возможности забрюшинного или внутрипочечного кровотечения. Кровотечение может быть как острым, так и хроническим и квалифицируется как угрожающее жизни состояние. При этом отмечаются абдоминальные боли, падение артериального давления, холодный пот, анемия, возможна гематурия и микроальбуминурия.
Как правило, для диагностики ангиомиолипом достаточно ультразвукового исследования, однако при подозрении на почечно-клеточную карциному показано проведение компьютерной и магнитно-резонансной томографии. Высокое содержание жировой ткани в ангиомиолипоме позволяет четко дифференцировать ее с карциномой. Однако при низком содержании жировой ткани в опухоли определить ее характер бывает сложно.
Кисты почек могут развиваться в любом отделе нефрона, бывают единичными или множественными, часто неотличимыми от кист при поликистозной болезни взрослого типа. Генез кистоза почек достаточно сложен. При туберозном склерозе основной причиной кист является гиперплазия клеток канальцевого эпителия, который при этом имеет специфические и уникальные, не встречающиеся при других видах кистоза черты: эозинофильность, крупное гиперхромное ядро. Эти клетки образуют скопления, нарушающие отток ультрафильтрата плазмы в нефроне, и вызывают расширение вышележащих отделов.
Почечные кисты выявляются при мутации как TSC1-, так и TSC2-генов. При мутации гена TSC1 кисты чаще солитарные, односторонние и выявляются реже, чем при мутации гена TSC2 [45].
Возраст выявления кист различен. Наиболее часто они диагностируются позже 10 лет, однако в ряде случаев поликистоз выявляется у детей раннего возраста, и даже при ультразвуковом исследовании плода.
Первыми клиническими симптомами кистоза почек могут быть боль в пояснице или гематурия. Исключением является рано выявленная поликистозная болезнь, которая может дебютировать артериальной гипертензией, обусловленной активацией ренин-ангиотензин-альдостероновой системы. Для постановки диагноза кистоза почек достаточно ультразвукового исследования. Внутривенная урография проводится в тех случаях, когда имеет место деформация чашечно-лоханочной системы с нарушением оттока мочи, присоединение инфекции. Часто поликистоз осложняется артериальной гипертензией, инфекцией мочевых путей, реже — кровотечением. Снижение функции почек, как правило, выявляется в 3–4-й декадах жизни. Прогрессированию почечной недостаточности способствуют тяжелые физические нагрузки, а также беременность. Однако в целом при туберозном склерозе терминальная хроническая почечная недостаточность (ХПН) возникает не часто.
Злокачественные опухоли при туберозном склерозе выявляются в 4,4% случаев, т. е. чаще, чем в популяции, с преобладанием у женщин (81%). Средний возраст их выявления — 29 лет. Билатеральное поражение встречается в 25% случаев. Наиболее часто диагностируется почечно-клеточная карцинома.
Летальность, связанная с патологией почек при туберозном склерозе, занимает второе место в структуре смертности после поражения центральной нервной системы. Наиболее часто причиной смерти становится ХПН. В 1996 г. F. Shillinger и R. Montagnac [48] описали 65 случаев туберозного склероза, все больные наблюдались в центрах гемодиализа Франции. Частота терминальной ХПН, по их данным, составила 1 : 100. Чаще она выявлялась у женщин (63,1%) и диагностировалась в среднем в 29-летнем возрасте. В половине случаев ХПН была первым симптомом манифестации туберозного склероза. Причинами ХПН при этом являлись: ангиомиолипоматоз в 23% случаев, поликистоз — в 18,5%, сочетание кист и ангиомиолипом — в 53%, злокачественные опухоли почек — в 13,8% случаев. ХПН при туберозном склерозе связана со сдавлением функционирующей ткани опухолевыми массами и/или кистами, а также с частичной или тотальной нефрэктомией (при операциях по поводу осложненных ангиомиолипом), что ведет к гиперфильтрации в сохранных нефронах и возникновению вследствие этого гломерулосклероза [48].
Трансплантация при туберозном склерозе, как правило, дает хорошие результаты (из 20 больных только у одного был криз отторжения). Перед трансплантацией рекомендуется проводить бинефрэктомию — во избежание кровотечения или малигнизации в сохраненных почках [48].
Консервативное лечение осложнений со стороны почек при туберозном склерозе состоит в постоянном применении гипотензивных средств при наличии скрытой или явной артериальной гипертензии (при этом препаратами выбора являются ингибиторы АПФ или блокаторы рецепторов ангиотензина), коррекции нарушения кальциевого обмена. При кистозном процессе в почках оперативное лечение проводится редко и заключается в декомпрессии кист. Хирургические вмешательства при ангиомиолипомах более распространены, проводятся они при угрозе или начавшемся кровотечении, а также при подозрении на малигнизацию. При угрозе кровотечения возможна эмболизация артерии, частичная нефрэктомия или тотальная в случае обширного поражения почки кистами и ангиомиолипомами.
В настоящее время проводятся исследования процесса онкогенеза при туберозном склерозе. В последние годы появились сообщения об открытии фактора, названного m-TOR (mammalian-target of rapamycin), стимулирующего синтез белков, клеточный рост и пролиферацию клеток. При мутации генов TSC1 и TSC2 нарушается продукция кодируемых ими белков: туберина и гамартина, которые в комплексе друг с другом в норме угнетают активность m-TOR-фактора. При поломке этих генов происходит его активация, что ведет к увеличению размеров клеток, наблюдаемому в гамартомах и при гипертрофии миокарда, а также к ускоренной их пролиферации. Фактор m-TOR назван так в связи с супрессирующим воздействием на него рапамицина — препарата, относящегося к группе иммуносупрессоров, широко применяемых в трансплантологии для предупреждения криза отторжения. Эксперименты на животных по применению рапамицина дали обнадеживающие результаты в виде значительного уменьшения размеров ангиомиолипом. Описания применения рапамицина у людей немногочисленны. Препарат, в отличие от цитостатиков, применяемых в трансплантологии, не является нефротоксичным. В настоящее время планируется проведение мультицентровых клинических испытаний рапамицина (сиролимуса) при туберозном склерозе.
Существует ряд больных, у которых при верифицированном диагнозе туберозного склероза уже в младенческом возрасте выявляется тяжелый поликистоз, клинически идентичный аутосомно-доминантной поликистозной болезни (АДПКБ). При этом состоянии предполагается мутация двух генов — TSC2 и PKD1, которые на коротком плече 16-й хромосомы прилежат друг к другу [15]. В основе этого синдрома лежит делеция, повреждающая оба гена. На основании сопоставления клинических данных и генетических исследований J. R. Sampson и соавторов [45] делают вывод, что нерезко выраженный кистоз при туберозном склерозе может быть связан с небольшой мутацией гена ТSС1 или ТSС2 без вовлечения гена PKD1, в то время как тяжелая и рано манифестирующая поликистозная болезнь при туберозном склерозе обусловлена сочетанной мутацией двух генов. В почках больных с сочетанным поражением генов туберозного склероза и АДПКБ наблюдаются морфологические черты обоих заболеваний: множественные ангиомиолипомы, кисты с типичным для туберозного склероза эозинофильным и гиперплазированным эпителием, внутриклубочковые поражения, а также характерные для АДПКБ крупные кисты, выстланные уплощенным эпителием. Причины раннего дебюта поликистозной болезни при туберозном склерозе на настоящий момент до конца не выяснены. Возможно, он связан с полной инактивацией гена PKD1, в то время как дебют АДПКБ в старшем возрасте объясняется сохранением некоторой активности гена PKD.
При наличии раннего дебюта поликистозной болезни при туберозном склерозе необходимо своевременное проведение суточного мониторирования артериального давления, определение уровня ренина и ангиотензина плазмы для назначения патогенетического лечения гипертонии ингибиторами АПФ, позволяющего отдалить формирование ХПН.
Литература
- Дорофеева М. Ю. Туберозный склероз у детей//Российский вестник перинатологии и педиатрии. 2001. № 4. С. 33-41.
- Калинин В. В. Новый антиэпилептический препарат Топамакс//Российский медицинский журнал. 2002. № 10. С. 1-9.
- Никанорова М. Ю., Белоусова Е. Д., Ермаков А. Ю., Перминов В. С. Фармакорезистентные эпилепсии у детей//Российский медицинский журнал. 2002. № 2. С. 43-45.
- Никанорова М. Ю., Ермаков А. Ю., Белоусова Е. Д, Достижения и проблемы фармакотерапии эпилепсий у детей//Российский вестник перинатальной педиатрии. 2002. № 5. С. 23-27.
- Никанорова М. Ю., Ермаков А. Ю., Горчханова З. К. Топирамат в лечении эпилепсии у детей (обзор литературы)//Фарматека. 2003. 4: 67. С. 23-25.
- Abad C., Trillo M., Olalla E. et al. Cardiac rhabdomyoma and tuberous sclerosis. Survival after the surgical resection of the cardiac tumor. Rev Esp Cardiol. 1991; 44: 4: 280-282.
- Abou-Khalif B. and the Topiramate YOL Study Group. Topiramate in the Long-Term Management of Refractory Epilepsy. Epilepsia. 2000; 41: 1: 72-76.
- Ben-Menachem E. Preliminary efficacy of levetiracetam in monotherapy. Epileptic Disord. 2003; 5: 51-55.
- Berkenblit R., Spindola F. H., Frater R. W., Fish B. B., Glickstein J. S. MRI in the evaluation and management of a newborn infant with cardiac rhabdomyoma. Ann Thorac Surg. 1997; 63: 5: 1475-1477.
- Bolton P. Intellectual and Cognitive Impairments. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003; 77-90.
- Bosi G., Lintermans J. P., Pellegrino P. A. et al. The natural hystory of cardiac rhabdomyoma with and without tuberous sclerosis. Acta Paediatr. 1996; 85: 928-931.
- Bosio M., Vitali G. M., Pandolfi M., Pastori P. Cardiac rhabdomyoma in tuberous sclerosis. A report of five cases and review of the literature. Minerva Pediatr. 1992; 44: 6: 305-311.
- Bourgeois B. Pediatric epilepsy Syndromes and their Surgical Treatment. Eds. I. Tuxhorn et al. London. 1997; 99-103.
- Brodie M. J., French J. A. Role of levetiracetam in the treatment of epilepsy. Epileptic Disord. 2003; 5: 65-72.
- Brook-Carter P. T., Peral B., Ward C. J. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease — a contiguous gene syndrome. Nat Genet. 1994; 8: 328-332.
- Cohen J. Levetiracetam monotherapy for primary generalized epilepsy. Seizure 2003; 12: 150-153.
- Curatolo P. Tuberous Sclerosis. In: Infantile Spasms and West Syndrome. Ed. by O. Dulac, H. T. Chugani, B. Dalla Bernandina. W. B. Saunders. Company Ltd, London, Philadelphia, Toronto, Sydney, Tokio. 1994; 192-202.
- Curatolo P., Cusmai R., Cortesi F. et al. Neuropsychiatric aspects of tuberous sclerosis. Annals of the New York Academy of Sciences. 1991; 615: 8-16.
- Curatolo P., Seri S. Seizures. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003; 46-77.
- Curatolo P., Verdecchia M. Neurological manifestations. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003; 26-45.
- Destuynder R., Menget A., Fromentin C. et al. Bourneville tuberous sclerosis manifested by prenatal finding of intracardiac tumors. Pediatrie (Bucur). 1992; 47: 4: 279-284.
- DiMario F. J. J., Diana D., Leopold H., Chameides L. Evolution of cardiac rhabdomyoma in tuberous sclerosis complex Clin Pediatr (Phila). 1996; 35: 615-619.
- Garnett W. R. Clinical Pharmacology of Topiramate: A Review. Epilepsia. 2000; 41: 1: 61-65.
- Gomez M. R. History of Tuberous Sclerosis Complex. In: Tuberous Sclerosis. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 3-9.
- Gomez M. R. Liver, Digestive Tract, Spleen, Arteries, Thymus and Lymphatics. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 228-239.
- Groves A. M., Fagg N. L., Cook A. C., Allan L. D. Cardiac tumors in intrauterine life. Arch Dis Child. 1992; 67: 10: 1189-1192.
- Hishitani T., Hoshino K., Ogava K. et al. Rapid enlargement of cardiac rhabdomyoma during corticotropin therapy for infantile spasms. Can J Cardiol. 1997; 13: 72-74.
- Hunt A. Psychiatric and Psychological Aspects. In: Tuberous Sclerosis. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 47-62.
- Inoue Y., Nemoto Y., Murata R. et al. CT and MR imaging of cerebral tuberous sclerosis. Brain & Development. 1998; 20: 209-221.
- Jozwiak S. Cardiac and Vascular Involvement. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003; 26-45.
- Kadar K., Buzas E., Geczi E., Lozsadi K. Rhabdomyomas as a first manifestation of childhood tuberous sclerosis. Orv Hetil. 1998; 139: 2013-2015.
- Kwiatkowski D. J., Reeve M. P., Cheadle J. P., Sampson J. R. Molecular Genetics. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003; 228-263.
- Lie J. T. Pulmonary Tuberous Sclerosis. In: Tuberous Sclerosis. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 207-217.
- Mair D. D., Edwards W. D., Seward J. B. Cardiac Manifestations. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 194-206.
- Nakayama K., Gu K., Yamauchi M. et al. Successful operation for isolated cardiac rhabdomyoma of the right atrium. Kyobu Geka. 1993; 46: 9: 803-806.
- Nir A., Tajik A. J., Freeman W. K., Seward J. B. et al. Tuberous sclerosis and cardiac rhabdomyoma. Am J Cardiol. 1995; 76: 5: 419-421.
- O’Callaghan F. J. K.,Clarke A. C., Joffe H. et al. Tuberous Sclerosis Complex and Wolff-Parkinson-White syndrome. Arch Dis Child. 1998; 78; 159-162.
- Ohtsuka Y., Ohmori I., Oka E. Long-Term Follow-Up of Childhood Epilepsy Associated with Tuberous Sclerosis. Epilepsia. 1998; 39: 1158-1163.
- Osborne J. P., Fryer A., Webb D. Epidemiology of Tuberous Sclerosis. Annals of the New York Academy of Sciences. 1991; 615: 125-128.
- Quek S. C., Yip W., Quek S. T. et al. Cardiac manifestatios in tuberous sclerosis: a 10-year rewiew. J Paediatr Child Health. 1998; 34: 624-628.
- Roach E. S., DiMario F. J., Kandt R. S., Northrup H. Tuberous Sclerosis Consensus Conference: Recommendations for Diagnostic Evaluation. Journal of Child Neurology. 1999; 14: 401-407.
- Robertson D. M. Ophthalmic findings. In: Tuberous Sclerosis. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 147-159.
- Rogers R. S., O’Connor W. J. Dermatologic Manifestations. In: Tuberous Sclerosis. Ed. M. Gomes, J. Sampson, V. Whittemore. New York; Oxford: Oxford University Press. 1999; 160-180.
- Sadi A. M., Toda T., Kiyuna M. et al. Cardiac rhabdomyoma of a neonate: an autopsy case report. Acta Paediatr Jpn. 1996; 38: 4: 361-364.
- Sampson J. R., Maheshwar M. M., Aspenwall R., Thomson P. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet. 1997; 61: 843-851.
- Shank R. P., Gardocki J. F., Streeter A. J., Maryanoff B. E. An Overview of the Preclinical Aspects of Topiramate: Pharmacology, Pharmacokinetics and Mechanism of Action. Epilepsia. 2000; 41: 3-9.
- Shepherd C. W., Scheithauer B. W., Gomez M. R. et al. Subependymal Giant Cell Astrocytoma: A Clinical, Pathological, and Flow Cytometric Study. Neurosurgery. 1991; 28: 864-868.
- Shillinger F., Montagnac R. Chronic renal failure and its treatment in tuberous sclerosis. Nephrol Dial Transplant. 1996; 11: 481-485.
- Shorvon S. D., Lowenthal A., Jans D. et al. Multicenter double-blind, randomized, placebo-controlled trial of levetiracetam as add-on therapy in patients with refractory partial seizures. Epilepsia. 2000; 41: 1179-1186.
- Stillwell T. J., Gomez M. R., Kelalis P. P. Renal lesions in tuberous sclerosis. J Urol. 1987; 138: 477-481.
- Torres O. A., Roach E. S., Delgado M. R. et al. Early Diagnosis of Subependymal Giant Cell Astrocytoma in Patients With Tuberous Sclerosis. Journal of Child Neurology. 1998; 13: 173-177.
- Van Veelen C. W. M., Hardus P., Verduin W. M. et al. Concentric contraction of visual field in patients with temporal lobe epilepsy using vigabatrin, long term results. Epilepsia. 2004; 41: 7: 136.
- Weig S. G., Pollack P. Carbamazepne-induced heart block in a child with tuberous sclerosis and cardiac rhabdomyoma: implications for evaluation and follow up. Ann Neurol. 1993; 34: 617-619.
М. Ю. Дорофеева, кандидат медицинских наук
О. С. Страхова, кандидат медицинских наук
О. В. Катышева
Э. К. Осипова, кандидат медицинских наук
О. И. Чумак
М. В. Добрынина
НИИПиДХ, Москва